Po co nam kontrola zmian w wyrobach medycznych ?
Kilka słów o kontroli zmian
Każda firma powinna wdrożyć swój własny system kontroli zmian oparty o wymagania prawa, norm czy własne doświadczenie. Ważne, aby nie pomijać tego procesu wewnątrz organizacji. Złożony system regulacyjny, ilość audytów oraz często liczba produkowanych wyrobów uniemożliwia zastosowania popularnego niegdyś systemu pod tytułem „ Prezes/Kierownik ma wszystko w głowie”.
Co to jest ta kontrola zmian?
W celu określenia jak wcześnie należy rozmawiać o zmianach lub kiedy możemy przestać je nadzorować warto przypomnieć sobie definicje „cyklu życia” czyli wszystkich faz życia wyrobu medycznego, od początkowej koncepcji do ostatecznego wycofania z eksploatacji i likwidacji.
Nadzorowanie zmian należy prowadzić na wszystkich etapach cyklu życia. Często zapominamy, że projekty wyrobów medycznych również żyją zgodnie z cyklem życia wyrobów i nie aktualizujemy / dodajemy zapisów dotyczących zmian. Nie chodzi tu aby zastąpić wersję pierwszą projektu a prowadzenie i nadzorowanie historii od pierwszej do końca życia wyrobu – coś na wzór Device History File.
W naszej ocenie najlepszą definicje ma niestety nie ISO a GMP. Definiuje ten proces jako :
kontrola zmian – formalny system, w ramach którego wykwalifikowani w odpowiednich dyscyplinach
nauki reprezentanci przeglądają proponowane lub faktyczne zmiany, które mogą wpłynąć na status
walidacji obiektów, systemów, urządzeń lub procesów; jego celem jest określenie potrzeby działań
zapewniających i dokumentujących utrzymanie systemu w stanie zwalidowanym;
Co może uruchomić ten proces ?
Każda firma ze względu na swoją działalność ( może produkować nie tylko wyroby medyczne dlatego może podlegać dodatkowym regulacjom) oraz ze względu na dany rodzaj wyrobów może zidentyfikować inne powody uruchomienia kontroli zmian. Przykładowe to:
- Zmiany organizacyjne, normatywne lub zmiany prawne ( najlepszy przykład ostatnich miesięcy to MDR)
- Zmiany w wyrobie ze względu na rosnące wymagania klientów lub trendy branżowe
- Zmiany w metodach produkcyjnych lub badań
- Ulepszanie lub wymiana parku maszynowego lub sprzętu laboratoryjnego ( należy wtedy zwrócić uwagę na walidację procesów czy metod badania)
- Dodanie lub poprawa jakości pomieszczeń/obszarów wytwarzania
- Obserwacje audytowe/inspekcyjne
- Zmiany wynikające z BHP
- Zmiany dostawców i usługodawców
- Nowe surowce lub dodanie kolejnego ( uwaga na biokompatybilność https://bmct.pl/pl/surowce-biokompatybilnosc-a-wyroby-medyczne/ )
- Zalecenia Organów Nadzoru
- Zmiany w etykietach, opakowaniach czy instrukcjach użycia
- Zmiany w elektronicznych systemach ( ERP, eQMS, LIMS itp.)
Niektóre zmiany wymagają również powiadomienia Jednostki Notyfikowanej ( czasem nawet jej akceptację) oraz Regulatora Rynku. Również nasz odbiorca ( hurtownia, dystrybutor lub pacjent ) powinien wiedzieć o zmianach które mogą wpłynąć np.: na bezpieczeństwo wyrobu lub sposób jego używania.
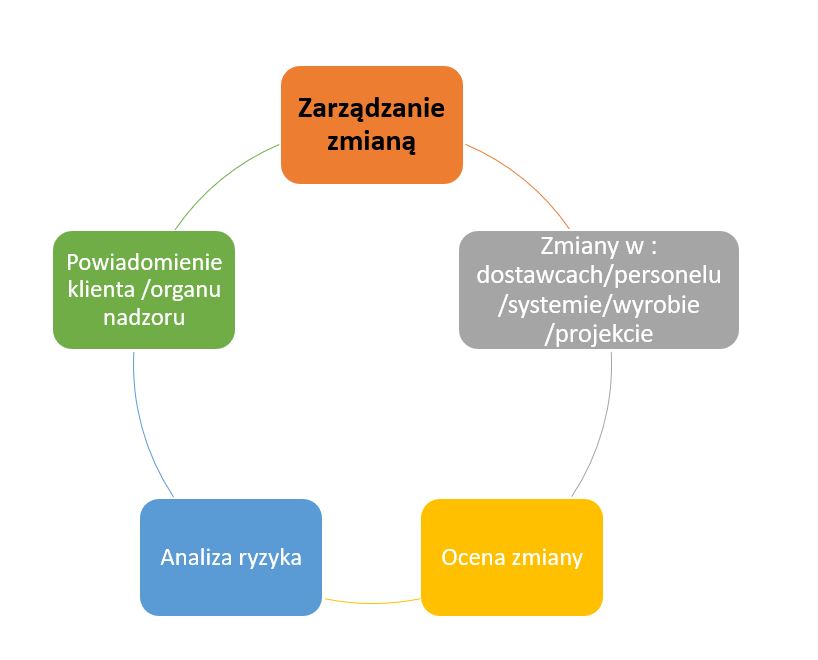
Jak prowadzimy zapisy ze zmian ?
Znów nie ma jednej odpowiedzi. Rejestry, arkusze, formularze – każde rozwiązanie jest dobre jeżeli zostało opisane w Państwa QMS i pozwala nie tylko zapisać co zmieniliśmy ale ocenić np.: stopień wdrożenia zmian albo skuteczność/efektywność wdrożenia w czasie. Wiele firm posiada już zintegrowane systemy elektroniczne gdzie nadzorują te procesy bez użycia zapisów papierowych.
Po co nadzorować zmiany ?
Oczywiście każdy odpowie, że robimy to ponieważ jest to wymóg norm, prawa itp. Kontrola zmian ma jedną może nie oczywistą zaletę. Rotacja pracownicza powoduje, że ludzie awansują lub zmieniają organizację a wyrób jest na rynku. Często musimy po latach zrozumieć dlaczego używamy teraz surowca X skoro w projekcie widniał Y lub dlaczego przestaliśmy badać np.: pH w naszym wyrobie. Zapisy kontroli zmian mogą nam odpowiedzieć na nasze pytanie i oszczędzić nie tylko czas ale pieniądze gdyż nie musimy powtarzać np. badań.
Drugą zaletą jest tzw. problem po latach. Niektóre wyroby mają dość długi okres przydatności do użycia. Kiedy nastąpi problem na rynku możemy przeanalizować czy to wpływ którejkolwiek ze zmian lub sprawa niezwiązana z nimi.
Tak jak zaznaczono na początku ile firm, ilu specjalistów tyle pomysłów na ten proces. Ważne aby każdy go prowadził.

